
Familiární hypercholesterolemie: aktuality
Familial hypercholesterolemia: news
Familial hypercholesterolemia is a metabolic disease with extensive clinical implications for its carriers. It is caused by a mutation in 1 of the 3 genes necessary for the metabolism of the LDL cholesterol particle. Suspicion of familial hypercholesterolemia should be suspected with levels LDL cholesterol > 5 mmol/L without significant triglyceride elevation. A positive family history contributes significantly to the clinical diagnosis. Clinical signs of familial hypercholesterolemia are becoming less common. In practice, scoring systems have been used successfully to quantify clinical suspicion. Molecular genetic testing is not necessary for treatment, yet it can be beneficial for the patient. Treatment begins with diet and lifestyle changes, followed by pharmacological treatment. We have statins, ezetimibe, possibly in combination with other hypolipidemic drugs. Since year 2018, we can indicate treatment with PCSK-9 inhibitors in patients with familial hypercholesterolemia. This treatment is organized in the Czech Republic in the form of center treatment.
Keywords:
statins – ezetimibe – familial hypercholesterolemia – LDL cholesterol – monacolin K – PCSK-9 inhibitors
Authors:
Martina Vaclová; Michal Vrablík
Authors‘ workplace:
Centrum preventivní kardiologie, III. interní klinika – endokrinologie a metabolismu 1. LF UK a VFN v Praze
Published in:
AtheroRev 2021; 6(3): 163-167
Category:
Overview
Familiární hypercholesterolemie je metabolické onemocnění s rozsáhlými klinickými dopady na své nositele. Je zapříčiněna mutací v 1 ze 3 genů nezbytných pro metabolizmus LDL-cholesterolové částice. Podezření na familiární hypercholesterolemii bychom měli pojmout při hladině LDL-cholesterolu > 5 mmol/l bez výrazné elevace triglyceridů. Ke klinické diagnóze významně přispívá pozitivní rodinná anamnéza. Klinické známky familiární hypercholesterolemie jsou čím dále méně časté. V praxi se s úspěchem používají skórovací systémy, které umožňují kvantifikovat klinické podezření. Provedení molekulárně genetického vyšetření není k léčbě nezbytné, přesto může být pro pacienta přínosem. Léčba se neobejde bez diety a změn životního stylu, dále je zahajována léčba farmakologická. K dispozici máme statiny, ezetimib, eventuálně kombinace s jinými hypolipidemiky. Od roku 2018 můžeme u pacientů s familiární hypercholesterolemií indikovat léčbu PCSK9-inhibitory, tato léčba je v ČR organizována formou centrové léčby.
Klíčová slova:
familiární hypercholesterolemie – PCSK9-inhibitory – LDL-cholesterol – statiny – ezetimib – monakolin K
Úvod
Familiární hypercholesterolemie (FH) je nejčastějším monogenně dědičným metabolickým onemocněním člověka.
Biochemicky je charakterizována snížením katabolizmu LDL-cholesterolových částic, což vede k vysokým hladinám LDL-cholesterolu v plazmě a jeho dlouhodobému ukládání do stěn cév a jiných tkání. Nejdůležitějším klinickým důsledkem je předčasná manifestace aterosklerotického procesu v podobě infarktů myokardu, cévních mozkových příhod či aterosklerotického postižení tepen dolních končetin. Méně častou komplikací aterosklerózy mohou být stenóza aortální chlopně, ruptura aorty či viscerální ischemie.
FH je zapříčiněna mutací v 1 ze 3 genů potřebných při vazbě LDL-částice na LDL-receptor na povrchu hepatocytu. Mutace jsou přenášeny autosomálně dominantně (velmi vzácně i recesivně) [1]. Nejčastěji bývá příčinou mutace v genu pro LDL-receptor (přibližně u 90 % nemocných), na druhém místě v genu pro apolipoprotein B100 (přibližně u 5 % nemocných) a velmi vzácně je příčinou mutace v genu pro proprotein konvertázy subtilizin/kexin typu 9 – PCSK9 (< 1 % nemocných) [2]. Pro úplnost je nutno dodat, že u nemalé části klinicky jasných pacientů s FH není kauzální mutace odhalena nebo je nalezena mutace, jejíž přímý dopad není zcela objasněn.
Diagnostika FH
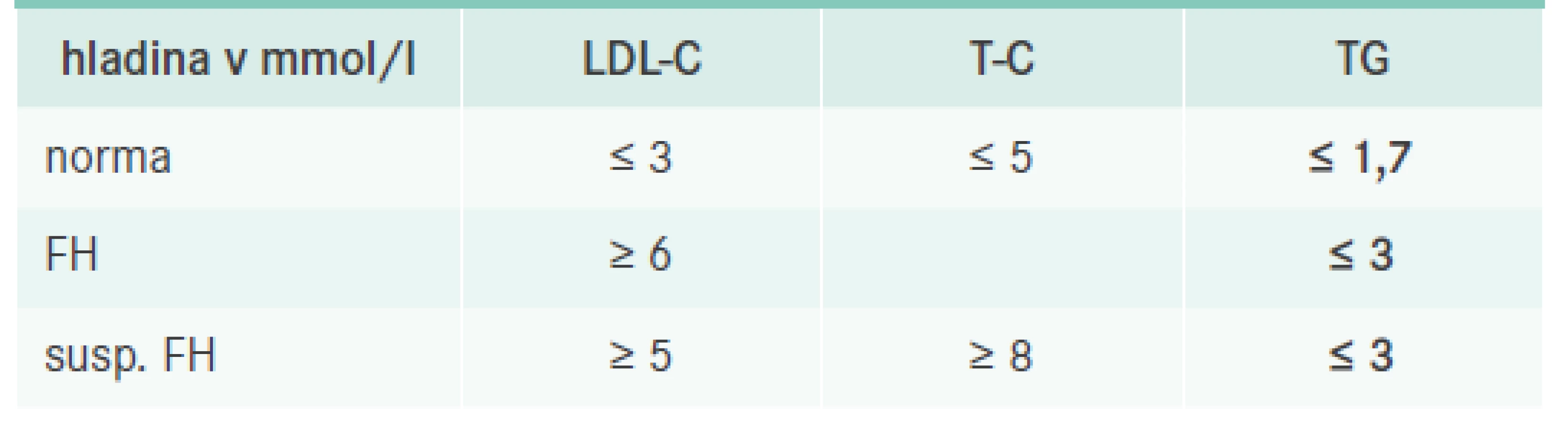
Diagnostika FH se odvíjí ve dvou rovinách. Nejdříve se jedná o diagnostiku klinickou, která je zásadní a opírá se o průkaz vysoké hladiny celkového (T-C) a LDL-cholesterolu (LDL-C) při normální či jen mírně elevované hladině triglyceridů (TG) spolu s pozitivní rodinnou anamnézou. Laboratorní testy by měly být provedeny za standardních podmínek, tedy po 10 hodinách lačnění v době, kdy se pacient cítí zdravý. Odběr by měl být potvrzen alespoň jedním kontrolním výsledkem s odstupem nejméně 6 týdnů a zároveň by měla být vyloučena možnost sekundární příčiny hypercholesterolemie (například gravidita, nefrotický syndrom, obstrukce žlučových cest, hypotyreóza, anorexie a vliv některých léků, jako jsou kortikosteroidy, pohlavní hormony včetně hormonální kontracepce). Pro rychlou orientaci si můžeme zapamatovat pouze několik čísel, u dospělého pojímáme podezření na FH vždy při hladinách LDL-C > 6 mmol/l nebo při hladině T-C > 8 mmol/l a současně LDL-C > 5 mmol/l (při hladině TG < 3 mmol/l), tab. 1. Pokud se jedná o dítě či adolescenta a/nebo pokud je pacient příbuzným již prokázaného pacienta s FH, pak je hladina potřebná k diagnostice FH nižší v závislosti na věku a stupni příbuznosti (u dětí bez FH příbuzných LDL-C > 4 mmol/l) [3]. Velký důraz se klade na kaskádový screening, který by měl být využit. U diagnostikovaného dítěte se vždy snažíme vyšetřit i jeho sourozence a rodiče, a naopak u dospělého pacienta s FH je vhodné vyšetřit jeho děti a sourozence, ev. rodiče, pokud je to možné (de facto se jedná o všechny prvostupňové příbuzné, u nichž je statistická pravděpodobnost výskytu onemocnění 50 %). Je velmi pravděpodobné, že kdyby se kaskádový screening striktně uplatňoval, odhalených pacientů s FH by bylo mnohem více.
Zároveň pátráme po pozitivní rodinné anamnéze. U velké části pacientů nacházíme nějakou formu předčasné manifestace kardiovaskulárních onemocnění (KVO). Za pozitivní se považuje výskyt KVO u prvostupňového příbuzného mužského pohlaví pod 55 let nebo u ženy pod 60 let věku. Přesto se stále častěji můžeme setkat i se situací, že je rodinná anamnéza buď zcela nebo částečně nevýtěžná (např. úmrtí rodiče v mladém věku na úraz či suicidiem, pacienti adoptovaní atd). V takovém případě bohužel přicházíme o cenný zdroj informací, který by nám v diagnostice FH pomohl. Ojedinělá není ani situace, kdy se nachází pozitivní rodinná anamnéza až u druhostupňových příbuzných a u prvostupňových nalezena není. Tento jev může být zapříčiněn, buď relativně nízkým věkem prvostupňových příbuzných (KVO nemají „zatím“) a/nebo jejich již delší dobu probíhající léčbou hypercholesterolemie, v takovém případě pak k manifestaci KVO nemusí dojít vůbec (což je skutečným cílem jejich léčby, na rozdíl od snížení hladiny cholesterolu, jak si mnozí pacienti bohužel myslí). Situaci s diagnostikou to však neulehčí.
Dalším ukazatelem, který může být nápomocný při stanovení diagnózy FH, jsou klinické příznaky. Klinické příznaky vyskytující se při déletrvající výrazné hypercholesterolemii nacházíme na víčcích očí v podobě xanthelasma palpebrarum, dále jako arcus lipoides corneae v podobě stříbřitě žlutavých kroužků nad okrajem duhovky oka (nemusí zabírat celý kruh) a také jako šlachové xantomy na šlachách především rukou a na šlachách Achillových. Příznaky hypercholesterolemie hledáme vždy u všech pacientů jak klinicky, tak i anamnesticky. V případě jejich nalezení je to velmi významná informace vedoucí nás k diagnostice familiární hypercholesterolemie. Prevalence klinických příznaků FH se stále snižuje. Vede k tomu běžně přístupná léčba hypercholesterolemie, z toho důvodu se klinické známky buď ani nevytvoří, nebo mohou s léčbou postupně vymizet dříve, než se pacient dostane do naší péče, v dnešní době je proto potřeba pátrat po klinických příznacích i anamnesticky, a to jak u rodiny, tak i u pacienta. Nezřídka se stává, že pacient podstoupí plastickou operaci víček, při které byla xanthelasmata odstraněna (někdy i opakovaně).
Jak vyplývá z výše uvedeného, klinická diagnostika FH není vždy zcela jednoduchá a jednoznačná. Z toho důvodu byly vyvinuty různé skórovací systémy, které mají za cíl diagnostiku usnadnit. Například se jedná o kritéria Simona Brooma nebo nizozemský systém Dutch Lipid Clinic Network Criteria (DLCN-kritéria) [4]. Obvykle se jedná o bodovací systém, ve kterém jsou body konkrétnímu pacientovi přiřazovány dle jeho rodinné a osobní anamnézy, fyzikálního vyšetření, dle nejvyšších výsledků hladiny LDL-cholesterolu v laboratorních výsledcích a ev. dle výsledků genetického vyšetření. V praxi se zdá nejkomplexnější systém DLCN-kritérií (tab. 2).
![Dutch Lipid Clinic Network Criteria pro diagnózu familiární hypercholesterolemie. Upraveno podle [4]](https://www.atheroreview.eu/media/cache/resolve/media_object_image_large/media/image_pdf/7f9c07c46f78cd4f9ddd2d2ca8034eb3.png)
Diagnóza FH je jistá při stavu skóre > 8 bodů, pravděpodobná při skóre 6–8 bodů a možná při skóre 3–5 bodů.
HDL-C– HDL-cholesterol ICHS – ischemická choroba srdeční LDL-C– LDL-cholesterol PCSK9 – proprotein konvertáza subtilizin/kexin typu 9
TG – triglyceridy
Dále můžeme, ale nemusíme, v diagnostice pokračovat genetickým vyšetřením, a to snahou o průkaz kauzální mutace. Genetické vyšetření přináší aspekty, se kterými je nutno počítat. Především jde o vyšetření, se kterým konkrétní pacient musí projevit písemný informovaný souhlas (bez souhlasu nelze k vyšetření přistoupit). Dále přináší nemalou finanční zátěž naštěstí jen jednorázovou. Případy, v nichž je genetické vyšetření potřeba opakovat, jsou velmi vzácné (v podstatě pouze při diskrepanci klinického a genetického vyšetření s podezřením na záměnu vzorků).
V neposlední řadě je potřeba zvážit, jakou informaci výsledek za vyšší cenu přinese a k jakému bude užitku. Z osobní praxe máme zkušenost, že kompliance k léčbě je u pacientů se známou mutací výrazně lepší než těch, kteří známou mutaci nemají a svou diagnózu familiární hypercholesterolemie často prezentují jako „vyšší cholesterol, který má dnes skoro každý“. Zároveň pacienti se známou mutací mají větší snahu a asi i argument pro vyšetření svých pokrevních příbuzných (především dětí a sourozenců). Do nedávné doby však genetické potvrzení klinické diagnózy familiární hypercholesterolemie nemělo přímý dopad na způsob a dostupnost léčby pro konkrétního pacienta. Léčba byla indikována zcela shodně, pokud pacient měl „pouze“ klinickou nebo i geneticky potvrzenou familiární hypercholesterolemii. Časy se však mění. V posledních 3 letech je k dispozici mimo jiné i biologická léčba hypercholesterolemie, konkrétně dva léky ze skupiny PCSK9-inhibitorů (alirokumab a evolokumab), která je indikována u pacientů s familiární hypercholesterolemií při nedostatečném snížení hladiny LDL-cholesterolu při maximální tolerované perorální léčbě (statinem, ev. v kombinaci s ezetimibem). V tomto případě lze s výhodou (naštěstí ne nezbytně) využít kritéria DLCN se zahrnutím genetické diagnózy familiární hypercholesterolemie.
Všeobjímající průnik technologií do našich životů můžeme také použít i při výpočtu bodových škál užívaných běžně k diagnóze familiární hypercholesterolemie (např. https://www.mdcalc.com/ nebo https://www.heartuk.org.uk).
Genetika
Jak bylo řečeno výše, výsledek molekulárně genetického vyšetření, ať již pozitivní či negativní, nemá přímý faktický vliv na léčbu pacienta.
Přednostně bychom měli uvažovat o genetickém vyšetření u pacientů, kteří mají velmi vysoké hodnoty LDL-C a je u nich podezření na homozygotní či smíšeně heterozygotní konstelaci mutací pro FH, dále pak u pacientů s těžkými nebo velmi časně se objevujícími klinickými známkami ze stejného důvodu. Stejně tak je výhodou, pokud je v jedné poradně léčeno více příbuzných, pak je možno otestovat jen jednoho z nich a ostatní pak testovat pouze na již odhalenou mutaci.
Genetické formy FH byly dříve děleny na FH heterozygotní a homozygotní dle průkazu 1 nebo obou mutovaných alel genu pro metabolizmus LDL-C (orientačně se jim přisuzovala i odpovídající hladina LDL-C). S přibývajícími poznatky v oboru molekulární biologie a přibývajícím množstvím geneticky vyšetřených pacientů s klinickou diagnózou FH však dochází k roztříštění tohoto zavedeného schématu. Geneticky se tedy může jednat o heterozygoty, homozygoty (s 1 totožnou mutovanou alelou) nebo o tzv. smíšené heterozygoty, kteří mají 2, avšak různé mutace alel stejného, či dokonce odlišného genu pro metabolizmus cholesterolu. Vzhledem k tomu, že sama přítomnost některé z mutací neříká nic o tíži klinického onemocnění ani o výši hladiny LDL-C v krvi, je nejlepší vyjadřovat výsledky genetického vyšetření pouze jako přítomnost dané mutované alely či alel s vysvětlením její klinické významnosti (pokud je známa) [5]. Není třeba se snažit je kategorizovat do jednotlivých podskupin. Je známo, že i jednotliví členové jedné rodiny nesoucí stejnou kauzální mutaci mohou mít různou tíži jak laboratorních, tak i klinických projevů. Závisí tedy jistě i na vlivu jiných genů a náš pohled na FH jako monogenně dědičné onemocnění se postupně, ale jistě mění na klinickou FH zapříčiněnou více či méně polygenně [6,7]. Podrobně se molekulárně genetickou diagnostikou u FH zabývá článek Mgr. Lukáše Tichého v tomto čísle AtheroReview.
Léčba: kdy, čím, jak?
Dietetická opatření a úprava životního stylu jsou nezpochybnitelným minimem, které pro svůj zdravotní stav může pacient udělat. Je potřeba si ale uvědomit, že zásadní je především trvání těchto opatření a pacientova celoživotní adherence k nim. Z toho důvodu preferujeme, ne imperativní, ale opakované vysvětlování vhodné diety se snahou o postupné zařazování změn, s tlakem na velmi dobrý životní styl po dobu běžného všedního života, ale s přípustným uvolněním diety ve dnech výjimečných a svátečních.
Naopak opatření k ukončení kouření by měla pro lékaře i pacienta být imperativem a zcela nezbytnou součástí léčby pacienta s familiární hypercholesterolemií, a to s využitím všech dostupných možností, vysvětlení, protikuřácké intervence, včetně konzultace v centru pro léčbu závislosti na tabáku, rozpisu nikotinových náhrad nebo eventuálního předpisu vareniklinu (např. www.bezcigaret.cz, www.slzt.cz).
Medikamentózní léčba FH je indikována u dospělých pacientů v okamžiku stanovení diagnózy. U většiny pacientů to nečiní obtíže, je nutno si uvědomit a zdůraznit jim, že vysoká hladina cholesterolu trvá od narození, a proto měl cholesterol již mnoho let možnost se jim „usazovat do cév“, ev. lze nastínit, že jejich „cévy jsou starší než zbytek jejich těla a kalendářní věk“ (pravděpodobně budou omezujícím faktorem jejich dožití).
U pacientů odhalených již v dětském věku zahajujeme léčbu nejpozději při dosažení dospělosti. V některých indikovaných případech dříve. Obvykle se v dětském věku zahajuje léčba pouze u homozygotů, u chlapců při obzvláště pozitivní rodinné anamnéze nebo při kumulaci významných rizikových faktorů. Nejčastěji je léčba zahájena ve věku 8–10 let u chlapců nebo později v adolescenci.
První volbou v léčbě familiární hypercholesterolemie jsou vysoce účinné statiny (atorvastatin a rosuvastatin) [8]. Dávku volíme individuálně, jak s přihlédnutím ke každému pacientovi, tak i k vlastní klinické zkušenosti. Lze začínat nižší dávkou 10 či 20 mg/den s úmyslem dávku navyšovat postupně až k dosažení cílové hodnoty LDL-C (tab. 3). Obzvláště vhodný je tento postup u starších pacientů, u pacientů s předchozí zkušeností s nežádoucími účinky či u pacientů s nižšími vstupními hodnotami, u kterých lze předpokládat dostatečnou účinnost i nižších a středních dávek léku. Naopak zahájení léčby maximálními dávkami léků lze doporučit u pacientů mladých, dosud bez zkušenosti s léčbou a nežádoucími účinky statinů či u pacientů s vysokými vstupními hladinami cholesterolu, u nichž lze předpokládat, že střední dávky statinů nebudou dostatečné. U obou postupů lze dosáhnout velmi dobrých výsledků za předpokladu, že s pacientem pracujeme citlivě, vysvětlujeme a respektujeme v něm partnera hodného spolupráce.
![Cílové hladiny LDL-C. Upraveno podle [3]](https://www.atheroreview.eu/media/cache/resolve/media_object_image_large/media/image_pdf/987632f943c6f36875810e0cf8038399.png)
Cílem léčby by mělo být vždy dosažení cílové hodnoty LDL-C, v první řadě využitím maximální potřebné či maximálně tolerované dávky vysoce účinného statinu. Pokud to není možné, snažíme se využít i ostatní dostupné statiny v nejvyšší možné tolerované dávce (simvastatin, fluvastatin, lovastatin). V uvedených případech přidáváme k maximální (tolerované) dávce statinu ezetimib, pokud nebylo dosaženo cílové hladiny LDL-C.
V případech nouze lze užívat i nestandardní dávkování statinů v kombinaci s ezetimibem, nebo i kombinaci s fibrátem (např. v případě přítomné hypertriglyceridemie nebo diabetu). Sekvestranty žlučových kyselin jsou v klinické praxi využívány opravdu minimálně, jak pro jejich horší dostupnost na trhu, tak i pro jejich četné gastrointestinální nežádoucí účinky a určité nepohodlí při užívání.
U některých pacientů lze s výhodou použít i monakolin K, volně prodejný potravinový doplněk získávaný přírodní cestou z plísně Monascuc purpureus, která barví substrát (nejčastěji rýži) na červeno. Extrakt získaný touto cestou obsahuje především monakolin K, který je chemicky identický s lovastatinem. Užívá se v dávce 10 mg/den a jeho účinek je slabší než účinek běžně užívaných statinů, avšak v kombinaci s ezetimibem může být snížení hladiny LDL-C uspokojivé (u některých pacientů se statinovou intolerancí nebo pacientů alternativně smýšlejících také bohužel jediné možné).
PCSK9-inhibitory
V posledních 3 letech máme možnost volby biologické léčby preparáty ze skupiny PCSK9-inhibitorů (alirokumab a evolokumab). Tuto léčbu je možno u familiární hypercholesterolemie indikovat z prostředků veřejného zdravotního pojištění při splnění úhradových podmínek [9] nebo formou samoplátcovství.
Odesílání do PCSK9 center
Do PCSK9 center by měli být odesíláni pacienti s FH diagnostikovanou jako pravděpodobná nebo jistá (dle kritérií DLCN nejméně 6 bodů) nedosahující při maximální statinové léčbě (rosuvastatin 40 mg/den nebo atorvastatin 80 mg/den) hodnot LDL-cholesterolu < 3,1 mmol/l, dále pak pacienti v sekundární prevenci KVO (s nebo i bez FH) při maximální statinové léčbě nedosahující hodnot LDL-C < 2,5 mmol/l.
Pokud má pacient parciální či úplnou intoleranci statinů, pak je nezbytně nutné, aby byly vyzkoušeny nejméně atorvastatin, rosuvastatin a ještě jeden jiný statin a tato skutečnost byla v dokumentaci precizně (včetně časových údajů a typu nežádoucích účinků) zaznamenána a dodána spolu s pacientem. V rámci provozu center ani vzhledem ke spolupráci s konkrétním pacientem není reálné ani etické v případě nedostatku informací opětovně všechny statiny zkoušet. V případě statinové intolerance se tedy k indikaci využívají hodnoty LDL-C při maximální tolerované statinové léčbě.
Ve všech případech může, ale nemusí být v kombinaci či monoterapii užit ezetimib. Pokud jsou dosažené výsledky alespoň o 20 % vyšší než hraniční hodnota, lze to komentovat tak, že by 20% snížení získané využitím ezetimibu nebylo dostatečné. Pak využití ezetimibu v medikaci není k úhradě PCSK9 podmínkou.
Dále považujeme za vhodné pacienta o zamýšlené léčbě injekčním preparátem před odesláním do centra informovat, situaci mu vysvětlit a uvážit, zda je aplikace schopen, i když jsou PCSK9-inhibitory vyráběny v předplněných perech a jejich aplikace je extrémně nenáročná a velmi dobře tolerovaná.
Závěr
Poznatky o FH, molekulárně genetická diagnostika i léčba postupují mílovými kroky kupředu. Přesto zůstává mnoho otazníků a FH je i nadále velmi zajímavým onemocněním, klinicky důležitým s rozsáhlými možnostmi jak na poli studia, diagnostiky tak i léčby. Pozitivní je, že informovanost laické populace i její zájem o vlastní zdraví výrazně narůstá. A tak je radostí s takovými pacienty pracovat.
MUDr. Martina Vaclová, PhD. | vaclova.martina@seznam.cz | www.int3.lf1.cuni.cz
Doručeno do redakce | Doručené do redakcie | Received 2. 7. 2021
Přijato po recenzi | Prijaté po recenzii | Accepted 21. 8. 2021
Sources
- Garcia CK, Wilund K, Arca M et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001; 292(5520): 1394–1398. Dostupné z DOI: <http://dx.doi.org/10.1126/science.1060458>.
- Češka, R. Familiární hypercholesterolemie. Praha: Triton 2015. ISBN 978–80–7387–843–6.
- Vrablík M, Piťha J, Blaha V et al. Stanovisko výboru ČSAT k doporučením ESC/EAS pro diagnostiku a léčbu dyslipidemií z roku 2019. AtheroRev 2019; 4(3): 19–30.
- van Aalst-Cohen ES, Jansen AC, Tanck MW et al. Diagnosing familial hypercholesterolaemia: the relevance of genetic testing. Eur Heart J 2006; 27(18): 2240– 2246. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehl113>.
- Brandts J, Dharmayat KI, Ray KK et al. Familial hypercholesterolemia: Is it time to separate monogenic from polygenic familial hypercholesterolemia? Curr Opin Lipidol 2020 Jun;31(3):111–118. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0000000000000675>.
- Hubacek JA, Adamkova V, Lanska V et al. Polygenic hypercholesterolemia: examples of GWAS results and their replication in the Czech-Slavonic population. Physiol Res 2017; 66(Suppl 1): S101–S111. Dostupné z DOI: Dostupné z DOI: <http://dx.doi.org/10.33549/physiolres.933580>.
- Tada H, Kawashiri MA, Nomura A et al. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J Clin Lipidol 2018; 12(6): 1436–1444. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacl.2018.08.006>.
- Mytilinaiou M, Kyrou I, Khan M et al. Familial hypercholesterolemia: New horizons for diagnosis and effective management. Front Pharmacol 2018 12; 9: 707. Dostupné z DOI: <http://dx.doi.org/10.3389/fphar.2018.00707>.
- Informace dostupné z WWW: <https://www.sukl.cz/sukl/prehledy-cen-a-uhrad-leciv>.
Labels
Angiology Diabetology Internal medicine Cardiology General practitioner for adultsArticle was published in
Athero Review

2021 Issue 3
Most read in this issue
- Nízký HDL-cholesterol: Jak to vlastně je?
- Familiární hypercholesterolemie: aktuality
- Kombinace statin a ezetimib: častěji v jedné tabletě a pro více pacientů
- Genetika familiární hypercholesterolemie: aktualizovaná kritéria pro interpretaci variant v genu LDLR