
Familiární hypercholesterolemie: klinické nálezy, molekulární genetika a diferenciální diagnostika
Familial hypercholesterolemia: clinical reports, molecular genetics and differential diagnosis
Recent studies have revealed the prevalence of familial hypercholesterolemia (FH) is approximately twice higher than previously estimated and, thus, the disease affects one in 250 persons from the general population. Therefore FH remains the most frequent inherited metabolic disorder. Due to the genetic defect LDL-cholesterol accumulates both in the plasma and tissues leading to premature and accelerated atherosclerosis. Untreated patients with FH might suffer myocardial infarction in the third or fourth decade, one third of these events being fatal. The disease is underdiagnosed and undertreated worldwide. The most effective way to identify FH patients is so called cascade screening in affected families. Early diagnosis and treatment significantly contribute to lowering of CVD risk. The treatment is based on lifestyle changes complemented with a maximal dose of statin. In case target LDL-cholesterol values are not reached a statin-ezetimibe combination is recommended. In the nearest future we will be able to use novel therapies (particularly PCSK9 inhibitors). These compounds together with MTP inhibitor lomitapide (indicated for homozygous and severe heterozygous FH) will bring up to 80 % of patients to their LDL-cholesterol targets.
Key words:
familial hypercholesterolemia – differential diagnosis – genetics – cardiovascular risk – cascade screening
:
Michal Vrablík 1; Lucie Schwarzová 1; Tomáš Freiberger 1; Lukáš Tichý 2; Richard Češka 1
:
Centrum preventivní kardiologie III. interní kliniky 1. LF UK a VFN, Praha
1; Genetická laboratoř, Centrum kardiovaskulární a transplantační chirurgie, Brno
2
:
AtheroRev 2016; 1(1): 19-27
:
Reviews
Nedávné studie ukázaly, že prevalence familiární hypercholesterolemie (FH) je přibližně 2násobná oproti dřívějším předpokladům, a onemocnění tak postihuje přibližně jednoho z 250 jedinců obecné populace. Jedná se tedy o nejčastější vrozené metabolické onemocnění vůbec. V důsledku geneticky podmíněné poruchy v metabolizmu LDL-cholesterolu dochází k jeho hromadění v cirkulaci a ve tkáních, což vede k předčasnému rozvoji aterosklerózy. Neléčení jedinci mohou mít infarkt myokardu ve 3. nebo 4. dekádě života, až v jedné třetině případů s fatálním koncem. Onemocnění je celosvětově nedostatečně diagnostikováno a léčeno. Logická je pozornost věnovaná včasné diagnostice FH s cílem zahájit léčbu co nejdříve a snížit tak u postižených jedinců riziko předčasné klinické manifestace ischemické choroby srdeční a předčasného úmrtí. Tomu napomáhá tzv. kaskádový screening mezi příbuznými pacientů s FH. Včasná diagnostika a zahájení léčby významně snižuje riziko postižených. Základem terapie FH jsou režimová opatření spolu s terapií maximální (tolerovanou) dávkou statinu. Není-li dosaženo cílových hodnot LDL-cholesterolu, doporučuje se přidání ezetimibu. V nejbližší budoucnosti budeme moci využít nových terapií (především PCSK9 inhibitorů). Jejich využití spolu s využitím blokátoru MTP lomitapidu (u homozygotů a těžkých heterozygotů FH) přivede k cílovým hodnotám až 80 % pacientů s FH.
Klíčová slova:
familiární hypercholesterolemie – diferenciální diagnostika – genetika – kardiovaskulární riziko – kaskádový screening
Úvod
Rozvoj kardiovaskulárních onemocnění v důsledku aterosklerózy je určován kombinací řady genetických faktorů a jejich dynamickou interakcí s vlivy vnějšího prostředí. Stejným způsobem je determinována dyslipidemie (DLP), která je důležitým rizikovým faktorem aterogeneze. Jednotlivé geny přitom mají na hladinu krevních lipidů jen malý účinek. Výjimkou jsou monogenní poruchy, při nichž defekt jednoho genu zásadním způsobem určuje výslednou podobu DLP. Modelovým příkladem monogenních DLP a zároveň nejzávažnější z nich je familiární hypercholesterolemie (FH). V klinické praxi se však mnohem častěji setkáváme s polygenními DLP, tedy poruchami metabolizmu lipidů, které mají genetický základ v mutacích či polymorfizmech více genů. Typickým příkladem metabolické poruchy determinované menšími odchylkami ve více genech je familiární kombinovaná hyperlipidemie (FKH), která je vůbec nejčastější primární DLP. Pro všechna polygenní onemocnění je charakteristický výrazně větší vliv zevního prostředí na výsledný fenotyp, nežli je tomu u monogenních poruch. Například určitá varianta genu pro apolipoprotein (apo) C vede k vzestupu triglyceridemie, ale za příznivých podmínek nedosáhne tato elevace horní hranice normálu. Budou-li však přítomny další faktory zvyšující hladinu sérových triglyceridů (obezita, léky, konzumace alkoholu, diabetes mellitus, další genetický faktor), můžeme u nositele takové genetické varianty nalézt manifestní hypertriglyceridemii [1]. Proto se někdy v literatuře uvádí termín DLP smíšené etiologie, jenž je vhodný právě pro situaci, v níž se DLP manifestuje u nositele genetické odchylky pouze při současném působení dalších faktorů. Polygenní DLP se zvýrazňují s věkem, což souvisí s větším výskytem nepříznivých faktorů zevního prostředí ve vyšším věku a samozřejmě i s celkovou délkou jejich působení.
Můžeme rozlišit DLP s izolovaným zvýšením cholesterolu, izolovaným zvýšením triglyceridů nebo kombinací obou (tab. 1). V následujícím přehledu bude největší pozornost věnována familiární hypercholesterolemii, která má největší význam z hlediska osudu svého nositele, je z genetického pohledu nejlépe definována a prozkoumána a v poslední době získáváme nové možnosti pro její léčbu.

Familiární hypercholesterolemie
FH se vyznačuje autosomálně dominantním typem dědičnosti a s frekvencí výskytu 1:200–250 ve většině bělošských populací představuje nejčastější vrozenou poruchu metabolizmu vůbec. Je charakterizována výrazným vzestupem LDL-cholesterolu v plazmě, jeho zvýšeným ukládáním ve tkáních, což se projevuje zejména šlachovými xantomy a tvorbou ateromatózních plátů v cévách, a s tím související předčasnou klinickou manifestací aterosklerózy, především ve formě ischemické choroby srdeční (ICHS). Riziko fatální nebo nefatální koronární příhody přesahuje 50 % u postižených mužů ve věku 50 let a 30 % u 60 letých žen [2,3]. Prognóza nemocných se dramaticky zlepšila na začátku 90. let minulého století po uvedení statinů na trh [3]. Většina pacientů s FH však stále zůstává nediagnostikována nebo je diagnóza stanovena až po první klinické manifestaci ICHS, která je až ve třetině případů fatální. Co nejčasnější diagnostika je tak velmi důležitá, neboť umožní včasné zahájení efektivní hypolipidemické léčby.
Etiopatogeneze FH
Již koncem 30. let 20. století postuloval Müller teorii, že familiární výskyt xantomů, vysokých hladin cholesterolu a infarktů myokardu je podmíněn defektem jednoho genu. V 60. letech pak Fredrickson et al demonstrovali, že fenotyp FH je spojen s poruchou metabolizmu lipoproteinů o nízké hustotě (LDL), a práci završili v 70. letech Goldstein s Brownem, kteří objevili LDL-receptor (LDL-R) a prokázali, že příčinou FH jsou mutace v genu kódujícím tento receptor [2]. Za svůj objev dostali v roce 1985 Nobelovu cenu. Vlivem porušené funkce LDL-R je významně sníženo vychytávání LDL-cholesterolu jaterními buňkami a dochází k jeho hromadění v cirkulaci, ve stěnách cév a dalších tkáních. Klinický fenotyp je výrazně těžší u homozygotů než u heterozygotů, protože u homozygotů nejsou v důsledku postižení obou alel LDL-receptory syntetizovány vůbec nebo jsou zcela nefunkční, případně je jejich funkce významně snížena. Studie na fibroblastech, které byly získány od homozygotních pacientů, ukázaly existenci mutací způsobujících pokles funkce LDL-R < 2 % („receptor negativní“ mutace), resp. na úroveň 2–25 % („receptor defektní“ mutace) [4]. Naproti tomu u heterozygotů zůstává jedna alela plně funkční a aktivita LDL-R je tak alespoň 50%. Homozygoti mají velmi vysoké koncentrace LDL-cholesterolu (> 13 mmol/l), tvoří se u nich kožní xantomy a manifestní ateroskleróza se vyvíjí již v prvních 2 dekádách života. S homozygotní FH je spojen i defekt genu LDL-RAP1 (LDL-receptor adaptor protein 1), označovaný také jako ARH, jehož defekt je příčinou autosomálně recesivní formy onemocnění (ARH, autosomálně recesivní hypercholesterolemie), která klinicky odpovídá homozygotní formě ADH a o níž pojednává samostatný odstavec tohoto článku [5].
Vzhledem k vzácnosti homozygotní formy (prevalence 1 : 106) se termín FH častěji užívá v souvislosti s heterozygoty. Koncentrace LDL-cholesterolu u heterozygotů jsou zvýšeny 2–3násobně a jsou více ovlivněny interakcí s dalšími geny i vlivy vnějšího prostředí, než je tomu u homozygotů. Stejně tak celkové kardiovaskulární riziko je u heterozygotů modulováno ostatními rizikovými faktory, zatímco u homozygotů se odvíjí téměř výhradně od koncentrací LDL-cholesterolu [6]. I u heterozygotů je ovšem role vnějších faktorů podstatně menší, než je tomu u jiných než monogenních forem DLP. Heterozygoti tvoří šlachové xantomy, které jsou pro FH patognomické, a poměrně časně také xantelazmata a arcus lipoides corneae, které patří k nespecifickým známkám nemoci. Manifestní ICHS se u nich vyvíjí od 3. dekády života a ve věku 50 let je postižena již více než polovina mužů s FH [2].
Část nemocných s klinickým fenotypem FH vykazuje normální aktivitu LDL-R. Bylo prokázáno, že za sníženou clearance LDL-cholesterolu z cirkulace je u těchto jedinců zodpovědná mutace R3527Q, resp. p.Arg3527Gln v genu pro apoB, a onemocnění dostalo název familiární defekt apoB-100 (FDB) [7]. ApoB-100 je jediným bílkovinným nosičem LDL-cholesterolu a mutace p.Arg3527Gln působí záměnu aminokyseliny argininu za glutamin v místě, které ovlivňuje vazbu apoB-100 k LDL-R. Později byly odhaleny další mutace v genu pro apoB, které vedou k FDB, ovšem ty se vyskytují velmi vzácně. Prevalence FDB je asi 1 : 800, přičemž nejvyšší je v oblasti střední Evropy. FDB a FH jsou klinicky nerozlišitelné, i když průměrné koncentrace LDL-cholesterolu jsou u heterozygotů FDB ve srovnání s heterozygoty FH nižší, šlachové xantomy méně frekventní a ke klinické manifestaci ICHS dochází později. Rozdíly jsou zřejmě dány nižší produkcí LDL-částic u pacientů s FDB v důsledku vyšší clearance remnantních partikulí přes neporušený LDL-R, protože tento proces je zprostředkován spíše apolipoproteinem E než apoB-100 [6].
U části pacientů s fenotypem autosomálně dominantní hypercholesterolemie (ADH) není přítomna mutace v genu pro LDL-R ani apo B. V takovém případě se hovoří o non-FH/non-FDB (non-LDL-R/non-APOB) hypercholesterolemii, případně FH3. Recentně byly u některých nemocných s FH3 detekovány mutace v genu PCSK9 (proprotein convertase subtilisin/kexin 9 – proprotein konvertáza subtilizin/kexin 9). Vyšetření větších souborů pacientů však ukázala, že tyto mutace mohou objasnit jen asi 2 % případů s fenotypem FH. Gen PCSK9 kóduje neurální apoptózou regulovanou konvertázu 1 (NARC1), jejíž role nebyla zatím plně objasněna. Zvýšená exprese PCSK9 má za následek posttranslační zvýšenou degradaci LDL-R. Mutace, které vedou ke zvýšené expresi PCSK9 („gain of function“ mutace) jsou asociovány s hypercholesterolemií a vyšším rizikem ICHS, zatímco mutace znamenající nižší koncentraci nebo dysfunkci NARC1 způsobují hypocholesterolemii a snižují riziko ICHS. PCSK9 se tak jeví být důležitým hráčem podílejícím se na výsledné hladině LDL-cholesterolu a různé varianty tohoto genu mohou být podstatnou měrou zodpovědné za její interindividuální variabilitu [8].
Stále více je v původním významu familiární hypercholesterolemie používán pojem autosomálně dominantní hypercholesterolemie (ADH), která v současné době zahrnuje:
- klasickou FH způsobenou mutacemi v genu pro LDL-R
- FDB s defektním genem pro apoB
- FH3 (resp. ADH3) s mutacemi v genu PCSK9
Epidemiologie FH
Nedávné studie založené na stanovení diagnózy FH podle modifikovaných široce užívaných kritérií (Dutch Lipid Network Criteria) u rozsáhlého vzorku více než 69 000 osob dánské populace [9] ukázaly, že prevalence FH je vyšší než 1 : 500, jak se dlouho uvádělo na základě práce Goldsteina et al ze 70. let minulého století [2]. Podle zmíněných recentních studií trpí familiární hypercholesterolemií přibližně 1 osoba z 250. Homozygotní FH je vzácná a výrazně závažnější forma onemocnění, postihující 1 ze 160 000 až 300 000 osob [10]. Přestože FH je velmi dobře definované onemocnění a přestože existuje účinná hypolipidemická léčba, většina pacientů s FH zůstává nediagnostikována nebo není adekvátně léčena [11]. Podle zmíněné dánské studie mají neléčení pacienti ve srovnání s obecnou populací 13krát vyšší riziko vzniku ICHS. Pozoruhodné je, že pacienti s FH, kteří léčeni jsou, mají pořád riziko vzniku ICHS 10krát zvýšené [3]. To svědčí o tom, že používaná léčba nevede u pacientů s FH k dostatečnému snížení hladin LDL-cholesterolu, respektive léčba zahájená v pozdějším věku není schopna zabránit manifestaci klinické komplikace aterosklerózy.
Diagnostika FH
Rodinná anamnéza
Autosomálně dominantní typ dědičnosti znamená přítomnost onemocnění u jednoho z rodičů, jednoho z prarodičů, poloviny sourozenců a poloviny potomků postiženého probanda. Vyšetření rodinných příslušníku je tedy velmi důležité, a to nejen z diagnostických důvodů, ale především proto, že může odhalit další jedince s FH, často ještě v asymptomatickém stadiu. V rodinné anamnéze je nutno pátrat po vysokém cholesterolu, šlachových xantomech a předčasném výskytu ICHS (u mužů do 55, u žen do 65 let věku), především u prvostupňových příbuzných. Vyšetření 4 příbuzných prvního stupně odhalí 2 nové pacienty, vyšetření 4 příbuzných druhého stupně dalšího jednoho pacienta s FH.
Klinické projevy
Kromě předčasného výskytu kardiovaskulárních příhod v osobní anamnéze je potřeba věnovat pozornost přítomnosti šlachových xantomů, které jsou pro FH patognomické. Vyšetření Achillovy šlachy a šlach dalších extenzorů by nemělo být opomenuto, i když absence šlachových xantomů rozhodně FH nevylučuje. Xantelazmata víček a arcus lipoides na rohovce patří k nespecifickým projevům choroby, ovšem jejich výskyt v nižších věkových skupinách může diagnózu FH podpořit. Klinické projevy obvykle chybějí u dětí a mladých dospělých. U pacientů s homozygotní formou FH se objevují i kožní xantomy.
Biochemické ukazatele
Důležitým diagnostickým pomocníkem jsou koncentrace celkového a především LDL-cholesterolu, přičemž by se mělo vycházet alespoň ze dvou měření. Nejpřesnější je použití hodnot specifických pro danou populaci, věk a pohlaví (tab. 2).
![Hodnoty 95. percentilu pro celkový a LDL-cholesterol specifické pro českou populaci podle věku a pohlaví. Upraveno podle [12]](https://www.atheroreview.eu/media/cache/resolve/media_object_image_large/media/image/02ce406e380eb37ef99bbed491163b74.png)
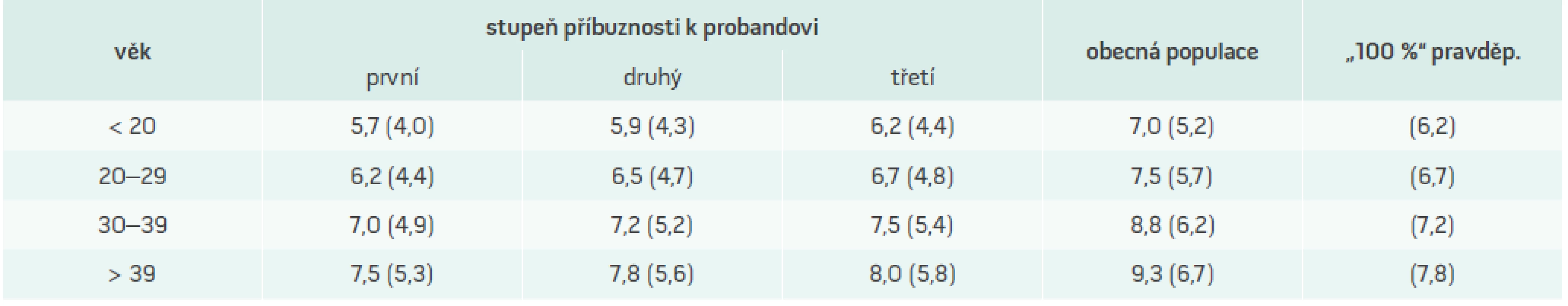
Diagnóza FH je zvažována při překročení hranice 95. percentilu za předpokladu, že byla vyloučena sekundární hypercholesterolemie. Koncentrace triglyceridů jsou typicky v normálních mezích, koncentrace HDL-cholesterolu jsou také normální. Samotná biochemická kritéria ovšem nejsou pro potvrzení diagnózy FH dostačující, a to vzhledem k překryvu s hodnotami nacházenými u části zdravé populace, resp. u osob s jinými formami hypercholesterolemie (FKH, těžší polygenní hypercholesterolemie apod). Kritéria vytvořená v rámci mezinárodního projektu MedPed zohledňují skutečnost, že příbuzní pacienta s již diagnostikovanou FH mají ve srovnání s jedinci obecné populace vyšší pravděpodobnost, že jsou rovněž postiženi, a proto pro ně byly vypočteny odlišné hraniční hodnoty rozhodné pro diagnózu, a to v závislosti na stupni příbuznosti (tab. 3). O projektu MedPed je pojednáno v jiném příspěvku časopisu.
Molekulárně genetická diagnostika
Určení příčinné mutace má význam pro zpřesnění diagnózy a může také pomoci u pacientů, u nichž nelze diagnózu jiným způsobem spolehlivě určit. Hlavní přínos molekulárně genetické diagnostiky ovšem spočívá v účinné pomoci při vyhledávání postižených rodinných příslušníků již diagnostikovaných pacientů s FH. Opakovaně bylo ukázáno, že 10–25 % příbuzných je zařazeno nesprávně, pokud je diagnostika založena pouze na koncentracích cholesterolu v plazmě [3]. V rodinách, u nichž je již mutace známa, je možno pomocí analýzy DNA rychle, spolehlivě a relativně levně určit postižené příbuzné. V současné době jsou známy tři geny, jejichž defekt vede k ADH.
Prvním krokem při genetické diagnostice pacientů s FH je screening několika mutací, o nichž je známo, že se v dané populaci vyskytují s největší frekvencí. Jako první je tedy v naší středoevropské populaci zjišťována přítomnost záměny p.Arg3527Gln v genu pro apolipoprotein B – příčina familiárně defektního apoB (FDB). Screening mutace může probíhat buď za použití restrikčních enzymů, při kterém hledaná mutace vnese či zruší místo střihu vhodně zvoleného enzymu. V případě záměny R3500Q (c.10580G>A) je použit enzym MspI, který v mutovaném řetězci DNA ztrácí své střihové místo a na výsledném agarózovém gelu pozorujeme delší fragment [13]. Modernější a rychlejší metodou je kvantitativní Real-time PCR (qPCR) s použitím fluorescenčně značených sond, specifických pro danou variantu DNA řetězce. Laserový detektor následně rozliší sekvenční varianty podle rozdílného fluorescenčního signálu. qPCR umožňuje i určit, zda se jedná o heterozygota (nejběžnější situace) či homozygota s mutací obou alel sledovaného genu.
Pokud zjistíme, že pacient nemá mutaci v genu pro apolipoprotein B, přistupujeme k vyšetření dalších genů zodpovědných za vznik FH, a to primárně genu pro LDL-receptor (LDL-R), popřípadě genu pro proprotein konvertázu subtilizin/kexin typu 9 (PCSK9). Opět je výhodné provést rychlý screening variant vyskytujících se s vysokou frekvencí v dané populaci, dnes nejčastěji prováděný pomocí qReal-time PCR. V naší populaci jsou nejčastějšími mutacemi v genu pro LDL-R záměny c.798T > A v exonu 5 a c.1775G > A v exonu 12 [14]. V případě nepřítomnosti některé z těchto mutací je následně gen sekvenován v celé délce 18 exonů. Klasická Sangerova sekvenace s použitím 4 specificky fluorescenčně značených dideoxynukleotidů, které po svém náhodném zařazení do rostoucího řetězce DNA brání navazování dalších nukleotidů, a následně umožňují laserovému detektoru odečítat délku takto vzniklých fragmentů a identitu posledního zařazeného nukleotidu, je proces časově náročný a pracný. Proto se v současné době mnoho diagnostických laboratoří snaží nahrazovat výše popsané zdlouhavé diagnostické analýzy rychlejší metodou – sekvenováním nové generace (next generation sequencing – NGS) [15]. Tato metoda umožňuje celogenomové sekvenování, exomové sekvenování i cílenou analýzu jednoho nebo několika lokusů v rámci genomu. NGS se v současnosti jeví jako nejvhodnější diagnostický nástroj pro rutinní vyšetřování známých kauzálních genů pro FH [16]. Během několika málo dní můžeme znát výsledek kompletního vyšetření několika genů pro desítky pacientů. Nejprve je DNA od každého pacienta amplifikována v místě vyšetřovaných lokusů a unikátně označena, poté jsou takto upravené vzorky více vyšetřovaných osob smíchány dohromady a osekvenovány. Nejčastěji využívanou technologií NGS je sekvenování při syntéze. Fluorescenčně značený reverzibilní terminátor vydává po své vazbě na rostoucí řetězec DNA signál, který je odečítán po každém přidání nukleotidu. Ten je poté odštěpen, aby byla umožněna vazba dalšího nukleotidu. Každá ze 4 bází nese unikátní fluorescenční značku, výsledkem je tedy sekvence odrážející pořadí nukleotidů v rostoucím fragmentu DNA. Tato data jsou následně vyhodnocována specializovaným softwarem, který zhodnotí množství čtení každého sledovaného lokusu s jednotlivými nalezenými sekvenčními variantami a na základě těchto dat odhalí přítomnost mutované alely. Celý diagnostický proces je díky moderním platformám, využívajícím popsané technologie, výrazně zkrácen a zpřesněn, zároveň umožňuje analýzu většího množství pacientů v jednom NGS běhu, a tím přináší i nezanedbatelnou celkovou finanční úsporu [17].
Komplexní diagnostická kritéria
Vedle MedPed kritérií (tab. 3) jsou pro diagnostiku FH nejčastěji využívána britská kritéria založená na datech z registru Simona Brooma nebo nizozemský skórovací systém (Dutch Lipid Network Criteria– DLNC). Britská kritéria ovšem nejsou pro naše podmínky příliš vhodná, protože se ve velké míře opírají o přítomnost šlachových xantomů, které se vyskytují u českých pacientů s FH mnohem méně často než u pacientů ve Velké Británii. Nizozemský systém je poměrně sofistikovaný (tab. 4) a zdá se použitelný i v podmínkách české populace. Jak Simon Broome register kritéria tak i DLNC považují průkaz kauzální mutace za dostačující pro stanovení definitivní diagnózy FH. Jak uvádíme v předchozím odstavci o molekulárně diagnostické diagnostice FH, ani ta nemůže sama o sobě jednoznačně posoudit přítomnost onemocnění. Máme nemocné negativně testované na všechny známé genetické příčiny FH s jednoznačným fenotypem onemocnění a na druhé straně osoby s prokázanou mutací v LDL-receptorovém genu bez fenotypových projevů. I proto nadále diagnóza FH včetně homozygotní formy zůstává klinickou a genetické vyšetření (jakkoli nesporně užitečné a důležité) představuje jedno z více kritérií choroby.

V diferenciální diagnostice významně zvýšených hladin celkového a LDL-cholesterolu musíme zvažovat i další formy geneticky podmíněných poruch manifestovaných izolovanou hypercholesterolemií. V každé z kategorií (rodinná anamnéza, osobní anamnéza, fyzikální vyšetření, laboratorní vyšetření, DNA analýza) je možno započítat pouze 1 bodovou hodnotu.
Autosomálně recesivní hypercholesterolemie
Fenotyp závažné izolované hypercholesterolemie, ovšem s odlišnou formou dědičnosti, sdílí vzácná autosomálně recesivní hypercholesterolemie (ARH). To je podmíněno mutacemi v genu pro LDL-receptor adaptorový protein 1 (LDL-RAP1). Adaptorový protein interaguje s cytoplazmatickým koncem LDL-receptoru a s klatrinem a je spoluzodpovědný za endocytózu LDL-receptorů. Jeho defekt vede k poruše internalizace LDL-receptoru s navázanou LDL-částicí, což je důvodem zvýšených koncentrací LDL-cholesterolu v cirkulaci. Množství LDL-R je normální, vazba LDL-cholesterolu na povrchu jaterních buněk dokonce zvýšená, ale změněna je distribuce LDL-R, který se nachází převážně na buněčné membráně a v menší míře v cytoplazmě. Postižený jedinec má defektní obě alely, zatímco jeho rodiče jsou zdraví přenašeči choroby. V rodinné anamnéze tak není patrný vertikální přenos onemocnění, což je výrazná odlišnost oproti FH, resp. ADH. Rodiče nesoucí poškozený gen mají 25% pravděpodobnost narození potomka s ARH [5]. Prevalence ARH je asi 1 : 5 × 106, více se vyskytuje u příbuzenských sňatků, které jsou ovšem v podmínkách České republiky nečetné. Pacienti s ARH odpovídají na léčbu statiny, zvýšená exprese LDL-R pravděpodobně částečně kompenzuje jeho defektní funkci, často je ovšem v terapii zapotřebí kombinace s LDL-aferézou.
Sitosterolemie
Přibližně stejně vzácná jako ARH je další autosomálně recesivní choroba popsaná poprvé rovněž na začátku 70. let minulého století, a to sitosterolemie. Koncentracemi cholesterolu a LDL-cholesterolu, xantomy i předčasnou klinickou manifestací aterosklerózy je velmi podobná homozygotní FH. Menší část pacientů je normocholesterolemická a nemá cévní onemocnění [18]. Charakteristickým rysem jsou více než 50násobně zvýšené koncentrace necholesterolových sterolů v plazmě a jejich ukládání ve tkáních. Specifickým znakem může být mírná hemolýza, k níž dochází pravděpodobně v důsledku inkorporace rostlinných sterolů do membrány erytrocytů. Heterozygoti nejsou postiženi a mají normální hladiny cholesterolu. Příčinou onemocnění jsou mutace na obou alelách genu pro ABCG5 nebo ABCG8, což jsou ATP vázající transportní kazety (typu G), které jsou zodpovědné za eflux živočišných i rostlinných sterolů z buňky. Pacienti se sitosterolemií mají zvýšenou frakční absorpci dietních sterolů a porušenou sekreci sterolů do žluči, což vede ke hromadění všech sterolů v krvi i ve tkáních. Diagnosticky cenné je stanovení vysokých koncentrací rostlinných sterolů (sitosterolu, kampesterolu, stigmasterolu, cholestanu apod) v plazmě [19]. Pacienti prakticky nereagují na léčbu statiny, zato přesvědčivě reagují na dietní restrikci cholesterolu a na sekvestranty žlučových kyselin či ezetimib [11].
Wolmanova choroba, nemoc z hromadění esterů cholesterolu
U pacientů se vzácnou autosomálně recesivní deficiencí kyselé lyzosomální lipázy (LAL; lysosomal acid lipase) dochází k masivní akumulaci esterů cholesterolu ve většině tkání. Lipidy se hromadí v lyzosomech, což vede ke zvýšené syntéze cholesterolu. V závislosti na věku začátku onemocnění a tíži příznaků je možno rozlišit Wolmanovu chorobu, která se manifestuje hned po narození a obvykle má fatální průběh před dosažením 1 roku věku, a nemoc ze střádání esterů cholesterolu (CESD; cholesteryl ester storage disease), která začíná v pozdějším věku a má mírnější průběh. Postižení kojenci neprospívají, mají hepatosplenomegalii, steatoreu a kalcifikace v nadledvinách, zatímco pacienti s CESD se prezentují často jen hepatomegalií, všichni mají výrazně zvýšený LDL-cholesterol a snížený HDL-cholesterol a velmi vysoké riziko předčasné klinické manifestace aterosklerózy [20]. Definitivní diagnóza spočívá v průkazu snížené aktivity LAL v lymfocytech. Lékem volby jsou statiny, ale existuje i specifická enzymová substituční terapie, která spočívá v dodávání defektního enzymu (sebelipase alfa). Tato terapie prošla úspěšně 3. fází klinického testování [21].
Polygenní hypercholesterolemie
Izolovaná primární hypercholesterolemie s rodinným výskytem, u které nalézáme obvykle nižší hodnoty celkového i LDL-cholesterolu než u FH, se nazývá polygenní hypercholesterolemie. Někteří nemocní s kumulací „nevýhodných“ alel zvyšujících LDL-cholesterol mohou však i při polygenní determinaci mít fenotypové projevy heterozgotní familiární hypercholesterolemie. Podobně jako u dalších polygenních hyperlipidemií je přesná genetická determinace polygenní hypercholesterolemie nejasná. Spekuluje se o vlivu polymorfizmů a mutací v genech pro apo E, CETP (cholesteryl ester transfer protein), jaterní a lipoproteinovou lipázu, LCAT (lecithin : cholesterol acyltransferase), PCSK9 a dalších, ale kombinace odchylek, která by jednoznačně vysvětlila fenotyp polygenní hypercholesterolemie není známa [22]. Mezi geny s největším účinkem patří apo E, jehož varianta apo E4 je asociována s vyššími hladinami cholesterolu než běžná varianta apo E3 [23].
Nemocní s polygenní hypercholesterolemií dobře reagují na režimová opatření a na terapii statiny, které lze v případě nedostatečného efektu nebo intolerance doplnit ezetimibem nebo pryskyřicemi [24].
Hlavní zásady léčby
Diagnostika a léčba pacientů s FH by měla být vedena ve spolupráci se specializovaným centrem. Není nutné pacienta do péče specialisty předávat, ale vždy při podezření na FH považujeme za vhodné pacienta konzultovat a eventuálně odeslat ke genetickému vyšetření a provedení rodinného (výše zmíněného) kaskádového screeningu na pracoviště specializovaného centra.
Léčbě pacientů s FH se věnuje celá řada recentních přehledů a byla přehledně a stručně formulována také ve Stanoviscích České společnosti pro aterosklerózu ke konsenzům European Atherosclerosis Society věnovaným této problematice [25,26]. Proto se v tomto přehledu omezíme pouze na schéma shrnující nejdůležitější body (schéma).
![Schéma. Strategie léčby pacienta s FH. Upraveno podle [11]](https://pl-master.mdcdn.cz/media/image/f4fdfd6f9502fddee416aa944827db00.png?version=1537797654)
Závěr
Genetické faktory mají zásadní význam pro určení povahy DLP, která je důležitým rizikovým faktorem pro rozvoj aterogeneze. U monogenních poruch převažuje účinek defektu jednoho genu a ostatní geny i faktory vnějšího prostředí mají pouze modifikující vliv na výsledný fenotyp onemocnění. Nejzávažnější monogenní DLP je familiární hypercholesterolemie podmíněná defektním genem pro LDL-receptor. U části pacientů s fenotypem FH tkví příčina nemoci v mutacích v genu pro apo B, resp. v genu PCSK9, u další části zodpovědný gen teprve čeká na své odhalení. Z klinického hlediska FH významně zvyšuje riziko aterotrombotických příhod a neléčená (nebo pozdě léčená) vede k předčasným úmrtím v důsledku klinické manifestace aterosklerózy. Vzhledem k tomu, že existuje účinná terapie, je včasné stanovení diagnózy a včasné zahájení léčby kriticky důležité. Možnosti léčby založené na režimových opatřeních v kombinaci s maximální (tolerovanou) dávkou statinu případně doplněné ezetimibem se v nejbližší budoucnosti obohatí o nové terapie. Nejblíže klinickému použití jsou inhibitory PCSK9, které spolu s blokátorem mikrosomálního triglyceridy transferujícího proteinu (MTP) lomitapidem (vyhrazeným pro případy homozygotní nebo těžké heterozygotní FH) umožní většímu počtu postižených dosáhnout cílových hodnot LDL-cholesterolu.
Práce byla podpořena grantem AZV 15–28277A.
Poděkování patří všem spolupracujícím lékařům (jejich seznam je uveden na http://athero.cz/cze/projekt-medped/sit-medped.php), koordinátorce Editě Firoňové, pracovníkům zajišťujícím molekulární diagnostiku, zejména doc. RNDr. Lence Fajkusové, CSc., Marii Plotěné a Mgr. Petře Zapletalové, společnosti Galén Symposion a Mgr. Haně Středové za pomoc s logistikou projektu a sponzorujícím společnostem Amgen, AOP Orphan, Krka, MSD, Pfizer a Sanofi.
doc. MUDr. Michal Vrablík, Ph.D.
michal.vrablik@lf1.cuni.cz
Centrum preventivní kardiologie III. interní kliniky 1. LF UK a VFN, Praha
www.vfn.cz
Doručeno do redakce 11. 1. 2016
Přijato po recenzi 28. 1. 2016
Sources
1. Lai CQ, Parnell LD, Ordovas JM. The APOA1/C3/A4/A5 gene cluster, lipid metabolism and cardiovascular disease risk. Curr Opin Lipidol 2005; 16(2): 153–166.
2. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver, CR, Beaudet AL, Sly WS et al. The metabolic and molecular bases of inherited disease. 4 voll. 8th ed. McGraw-Hill: New York 2001: 2863–2914. ISBN 978–0071163361.
3. Marks D, Thorogood M, Neil HA et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003; 168(1):1–14.
4. Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992; 1(6): 445–466.
5. Hegele RA. Monogenic dyslipidemias: window on determinants of plasma lipoprotein metabolism. Am J Hum Genet 2001; 69(6): 1161–1177.
6. Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest 2003; 111(12): 1795–1803.
7. Innerarity TL, Weisgraber KH, Arnold KS et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci USA 1987; 84(19): 6919–6923.
8. Cohen JC, Boerwinkle E, Mosley TH Jr et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354(12): 1264–1272.
9. Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 2012; 97(11): 3956–3964.
10. Cuchel M, Bruckert E, Ginsberg HN et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2012; 35(32): 2146–2157.
11. Nordestgaard BG, Chapman MJ, Humphries SE et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J 2013; 34(45):3478–3490a.
12. Šamánek M, Urbanová Z. Hodnoty cholesterolu a triglyceridů a jejich vývoj mezi druhým a sedmnáctým rokem. Čas Lék Čes 1997; 136(12): 380–385.
13. Hansen PS, Rudinger N, Tybjerg-Hansen A et al. Detection of the apoB-3500 mutation (glutamine for arginine) by gene amplification and cleavage with MspI. J Lipid Res 1991; 32(7): 1229–1233.
14. Tichý L, Freiberger T, Zapletalová P et al. The molecular basis of familial hypercholesterolemia in the Czech Republic: Spectrum of LDLR mutations and genotype-phenotype correlations. Atherosclerosis 2012; 223(2): 401–408.
15. Hartgers ML, Ray KK, Hovingh GK. New Approaches in Detection and Treatment of Familial Hypercholesterolemia. Curr Cardiol Rep 2015; 17(12): 109.
16. Hegele RA, Ban MR, Cao H et al. Targeted next-generation sequencing in monogenic dyslipidemias. Curr Op Lipidol 2015; 26(2): 103–113.
17. Vandrovcova J, Thomas ERA, Atanur SS et al. The use of next-generation sequencing in clinical diagnosis of famolial hypercholesterolemia. Genet Med 2013; 15(12): 948–957.
18. Hansel B, Carrie´ A, Brun-Druc N et al. Premature atherosclerosis is not systematic in phytosterolemic patients: severe hypercholesterolemia as a confounding factor in five subjects. Atherosclerosis 2014; 234(1):162–168.
19. Kidambi S, Patel SB. Sitosterolaemia: pathophysiology, clinical presentation and laboratory diagnosis. J Clin Pathol 2008; 61(5): 588–594.
20. Bernstein DL, Hulkova H, Bialer MG et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230–1243.
21. Burton BK, Balwani M, Feillet F et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med 2015; 373(11): 1010–1020.
22. Talmud PJ, Shah S, Whittall R et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 2013; 381(9874): 1293–1301.
23. Knoblauch H, Bauerfeind A, Krahenbuhl C et al. Common haplotypes in five genes influence genetic variance of LDL and HDL cholesterol in the general population. Hum Mol Genet 2002; 11(12): 1477–1485.
24. Soška V, Vaverková H, Vrablík M et al. Stanovisko výboru ČSAT k doporučením ESC/EAS pro diagnostiku a léčbu dyslipidemií z roku 2011. DMEV 2013; 16(1): 24–29.
25. Vrablík M, Freiberger T, Bláha V et al. Souhrn konsenzu panelu expertů European Atherosclerosis Society k otázce diagnostiky a klinickému vedení nemocných s familiární hypercholesterolemií. Pracovní skupina České společnosti pro aterosklerózu. Hypertenze a KV prevence 2015; 4(2): 44–48. Dostupné z WWW: http://www.hypertension.cz/sqlcache/hypertenze-02–2015.pdf.
26. Vrablík M, Češka R, Bláha V et al. Souhrn konsenzu panelu expertů European Atherosclerosis Society k otázce diagnostiky a klinickému vedení nemocných s homozygotní formou familiární hypercholesterolemie. Hypertenze a KV prevence 2015; 4(1): 54–56. Dostupné z WWW: http://www.hypertension.cz/sqlcache/csh-2015–1-web.pdf.
Labels
Angiology Diabetology Internal medicine Cardiology General practitioner for adultsArticle was published in
Athero Review

2016 Issue 1
Most read in this issue
- Familial hypercholesterolemia: clinical reports, molecular genetics and differential diagnosis
- PCSK9 inhibitors in the management of patients with high cardiovascular risk – effective treatment to reach
- Turn in hypercholesterolemia treatment – PCSK9 inhibitors. What we know about the alirocumab (product Praluent®) yet?
- Statin-associated myopathy: clinical guideline of Slovak Atherosclerosis Association and Czech Society for Atherosclerosis